アルツハイマー病: Alzheimer's disease
概要
以下は文献 1 のアルツハイマー病 Alzheimer's disease (AD) の説明である。記憶力の減衰などが原因で,日常生活に支障をきたす症状を認知症 dementia というが,AD は認知症の主な原因であり,患者数は非常に多い。認知症の診断基準は,DSM-5 などで定義されている。
|
アルツハイマー病は緩徐進行性の原発性脳変性疾患で全認知症患者の50%以上を占める。記憶障害を初発症状とし,見当識障害や失行,失認が生じる。初期にうつ症状を示すことがよくある。40-90 歳の間で発症することが多く,65 歳以下では早発性,65
歳以上では晩発性とされる。危険因子は加齢,女性,低教育歴,頭部外傷歴,遺伝性(apoE の ε4 多型)である。
肉眼病理上は全脳のびまん性萎縮を示す。組織学的には神経原線維変化(神経細胞内で Tau protein が異常リン酸化し蓄積したもの)が側頭葉内側の嗅内皮質に出現し,進行すると側頭,頭頂,後頭葉協会行きの大脳皮質にも出現,拡大する。重症度と相関し,健常人にはほとんどみられない。
Braak は神経原繊維出現頻度により Stage 1 - 6 に進行度を分けている。アルツハイマー病発症前段階の MCI (mild cognitive impairment) は Stage 2 - 3 に該当し,軽度アルツハイマー病は Stage 3 - 4,中等度アルツハイマー病は Stage 4 - 5 に該当する。一方,老人斑(アミロイド周囲に神経細胞などが集合したもの)はアルツハイマー病の初発所見とされるが,老人斑の量や範囲は重症度とは相関せず,健常老人でもみられる。 |
疫学調査: Epidemiological survey
> 65 歳以上人口の 13%,85 歳以上人口の 45% が AD を発症していると見積もられている(15)。
> 遺伝性 Familial AD の割合は 5% 程度と低く,ほとんどが散発性 sporadic である(5I)。
: ごく少数が家族発生し,プレセニリン 1 遺伝子などが関与する(1)。
: 3 つの遺伝子が familial AD と関連する。Amyloid precursor protein, presenilin, apolipoprotein E (7).
> 新潟県糸魚川での調査。65 歳以上: 6.2%が認知症, AD 4.0%, vascular dimentia 1.2%, others 0.3% (4R)。
: 6.2% は他の調査よりやや高いが,糸魚川は年齢層が高く,地方であるのでそのためかもしれない(4D)。
原因
アルツハイマー病の病態は多様であり,全ての病態を説明できる原因というものはない(11)。遺伝性ADならば原因遺伝子の同定は可能であるが,とくに散発性の AD において原因の特定は進んでいない(5I,7)。
アミロイドβ の蓄積と過剰リン酸化タウ hyperphosphorylation of Tau protein の蓄積は多くの病態で観察される現象で,有力な原因として考えられている。ただし,これは認知症の程度と相関する,またはしないの両方の報告がある。
また,アルツハイマー病患者の脳ではアセチルコリン acetylcholine が不足しており,分解酵素アセチルコリンエステラーゼの阻害剤フィゾスチグミン,タクリン,ドネパジルなどが治療薬として用いられている(13)。
原因として提唱されている現象
- アミロイドβの蓄積 amyloidgenesis(5I, 7)
-
Hyperphosphorylation of Tau(5I, 11)
- Neuronal cytoskeletal degeneration hypothesis(7)
- カルシウム代謝の異常 Disruption of Ca homeostasis(5I)
- エネルギー代謝の異常 Energetic failure(5I)
- ミトコンドリア機能不全(7)
- 酸化ストレス Oxidative stress (5I)
- 金属イオン(7)
- Cholineric hypothesis(7, 10I)
- 炎症 Inflammation(7)
家族性ADで報告されている変異
Amyloid precursor protein (APP)
> 20以上の病原性変異がみつかっている(8)。
V717I London mutation, V717F Indiana mutation, K670D/M671L Swedish or APP mutation, E693G Arctic mutation.
Presenilin 1, 2 (PSEN1, 2)
> 家族性 AD は主に presenilin 遺伝子の変異により,これまでに 130 以上の変異が報告されている(8)。
> Presenilin の変異は,家族性 AD の 90% を占める(11)。
脳の形態的変化
Atrophy 萎縮
特定の部位で,神経細胞 neuron やシナプスが失われるとともに,β-アミロイドやNFTsの蓄積がみられるのが特徴。文献によって萎縮がみられるとする場所に違いがあるが,海馬 hippocampus や大脳皮質 cortex の萎縮は数多く報告されている。
> Entorhinal cortexが最も早く, わずかに遅れてhippocampus, amygdala, and parahippocampus (2).
> 順にHippocampus, cortex, amygdata and nucleus basalis of Meynert (8).
: Nucleus basalis of Meynert is a group of cholinergic nerve cells in the basal forebrain.
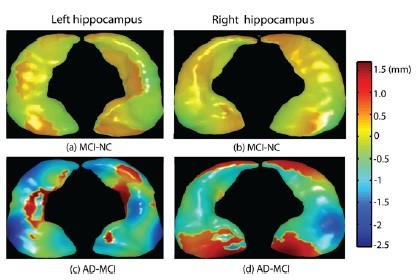
軽度認識傷害(MCI; mild cognitive impairment)およびアルツハイマー病(AD)でみられる海馬の萎縮(文献9より転載)。
たとえば左下のAD-MCIの青い部分は,MCI患者に比べてAD患者で2 mm前後の萎縮が起こっていることを示している。同様に,左上のパネルをみると,MCI患者ではNC(normal control)に比べてあまり萎縮が起こっていないこともわかる。
側脳室lateral ventricleでも同様の傾向がみられる。MCIとADは,これらの部位の容積よりも形に大きな違いが出る。
脳のグルコース代謝変化
代謝が低下する部位
以下の場所で,グルコース代謝の低下 hypometabolism が観察されている。多くは FDG-PET のデータであるが,PETは空間分解能の低いイメージング手法(4 - 5 mm)であり,あまり詳細な部位の特定はできない。
文献15では,グルコース代謝の低下するエリアは以下のようになっている。やや古い文献なので注意。
> 大脳皮質では,多くの部分でグルコース代謝が低下する(15R)。
: 頭頂葉 parietal lobe, 側頭葉 temporal lobe, 前頭葉 frontal lobe, 後頭葉 occipital lobe の皮質。
: 視床の値で標準化すると,後頭葉は有意差がなくなる。影響が小さいと言える。
> 感覚運動皮質 sensorimotor cortex は影響が小さい(15R)。
> 視床 thalamus では代謝は低下しない(15R)。
> 通常,グルコース代謝の低下は左右対称に観察される。ただし低下の程度は左右対称とは限らない(15I)。
Cortex 大脳皮質
- Posterior midline cortices of the parietal (precuneus) and posterior cingulate gyri (2R)
- Medial temporal cortices (2R).
- Primary cortex (2R).
- Bilateral parietal cortex (12R)
- Bilateral temporal cortex (12R)
Hippocampus 海馬 (2R)
The inferior parietal lobule (2R)
Posterolateral portions of the temporal lobe (2R)
The parental cingulate (14I)
The posterior cingulate (12R, 14I)
診断への応用
> PET で検出される低グルコース代謝は,認知機能テスト ADAS-cog や FAQ のスコア低下を予測できる(14)。
: つまり 実際に認知機能が低下する前に脳のグルコース代謝が低下するということ。
Others
> 脳の各部位で fMRI シグナルが同調して変化することを connectibity という。ADでは一般に相関が低下(6)。
: 記憶に関連する領域である海馬 hippocampus で,左右の相関が低下するのは有名である。
Animal models
Mouse
Tau models
> Longest human wt tauをneuronで発現するTg miceが最初のtau model。pre-tangle形成など(8)。
> のち,P301Lという変異の入ったtauを発現するモデルが作られ,広く用いられるようになった(8)。
> 他にもN279K, delta-K280, P301L, P301S, V337 and R406W のTg miceが作られている(8)。
APP models
> Human amyloid precursor protein 23 (APP23) transgenic mice (3I). Neuronal promoter.
> 6ヶ月齢ごろからAβをneocortex, hippocampusに蓄積。AD患者に似たcognitive declineを示す(3I)。
> Paw stimulationによるsomatosensory areaの血流増大が,25ヶ月齢でwt mouseより低い(3R)。
APPとtauの複合型
> JNPL3 strain: APP Tg strain Tg2576 と P301L tauをかけ合わせたもの(8)。
> pR5 strain: P301 tau strainにAβ42を注射したもの。症状が激しくなる(8)。
Secretase models
> APPのプロセシングを行うβ-,γ-secretaseの変異体もいる(8)。
ApoE models
> APOE遺伝子のε4 alleleは,アルツハイマー病のリスクファクター(8)。
> APP Tg miceをAPOE-/-にかけ合わせるとAβの沈着が抑えられるが,そこにApoE4を入れると増える(8)。
Axonal transport models
> 軸索の物質輸送を行う kinesin light chain のKlc+/- mouseはAPP異常により症状が出やすい(8)。
Rat
FAB rat
> Fe2+,Aβ42,グルタチオン合成阻害剤 buthionine-sulfoximine 注射でAD様の症状を示す(5R)。
> 多くのtransgenic modelがfamilial ADのモデルであるのに対し,これはsporadic ADのモデル(5R)。
> AβおよびNFTs蓄積,海馬や大脳皮質での神経細胞死,Morris water mazeスコア低下など(5R)。
> 前2者のみを注入しても症状はみられないことから,脳の酸化ストレス耐性の低下が発症に必要といえる(5D)。
References
- よくわかる脳 MRI 第 3 版, 秀潤社, 2013
- Johnson 2012a (Review). Brain imaging in Alzheimer disease. Cold Spring Harb Perspedt Med 2, a006213.
- Mueggler 2003a. Age-dependent impairment of somatosensory response in the amyloid precursor protein 23 transgenic mouse model of Alzheimer’s disease. J Neurosci 23, 8231-8236.
- Nakamura 2003a. Prevalence and predominance of Alzheimer's type dimentia in rural Japan. Phychogeriatrics 3, 97-103.
- Lecanu 2006a. Beta-amyloid and oxidative stress jointly induce neuronal death, amyloid deposits, gliosis, and memory impairment in the rat brain. Pharmacology 76, 19-33.
- Zhang 2010b (Review). Disease and the brain's dark energy. Nat Rev Neurol 6, 15-28.
- Tiiman 2013a (Review). The missing link in the amyloid cascade of Alzheimer's diseare - metal ions. Neurochem Int 62, 367-378.
- Gotz 2008a (Review). Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci 9, 532-544.
- Yang 2012a. CSF and brain structural imaging markers of the Alzheimer's pathological cascade. PLoS One 7, e47406.
- Kashiwaya 2000a. D-β-hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. PNAS 97, 5440-5444.
- Dong et al. 2012a (Review). Advances in the pathogenesis of Alzheimer's disease: a re-evaluation of amyloid cascade hypothesis. Transl Neurodegener 1, 18.
-
Arbizu et al. 2013a. Automated analysis of FDG PET as a tool for single-subject probabilistic
prediction and detection of Alzheimer's disease dimentia. Eur J Nucl Med Mol Imaging 40, 1394-1405.
-
河合良訓 監修 2005a. 脳単―ギリシャ語・ラテン語 (語源から覚える解剖学英単語集 (脳・神経編))
- Landau et al. 2011a. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Ageing 32, 1207-1218.
- Liu et al. 2013a. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9, 106-118.